Effects of Nitric Oxide Synthase Inhibition on Fiber-Type Composition, Mitochondrial Biogenesis, and SIRT1 Expression in Rat Skeletal Muscle.
Suwa, M; Nakano, H; Radak, Z; Kumagai, S
Journal of sports science & medicine
14
548-55
2015
Show Abstract
It was hypothesized that nitric oxide synthases (NOS) regulated SIRT1 expression and lead to a corresponding changes of contractile and metabolic properties in skeletal muscle. The purpose of the present study was to investigate the influence of long-term inhibition of nitric oxide synthases (NOS) on the fiber-type composition, metabolic regulators such as and silent information regulator of transcription 1 (SIRT1) and peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α), and components of mitochondrial biogenesis in the soleus and plantaris muscles of rats. Rats were assigned to two groups: control and NOS inhibitor (N (ω)-nitro-L-arginine methyl ester hydrochloride (L-NAME), ingested for 8 weeks in drinking water)-treated groups. The percentage of Type I fibers in the L-NAME group was significantly lower than that in the control group, and the percentage of Type IIA fibers was concomitantly higher in soleus muscle. In plantaris muscle, muscle fiber composition was not altered by L-NAME treatment. L-NAME treatment decreased the cytochrome C protein expression and activity of mitochondrial oxidative enzymes in the plantaris muscle but not in soleus muscle. NOS inhibition reduced the SIRT1 protein expression level in both the soleus and plantaris muscles, whereas it did not affect the PGC-1α protein expression. L-NAME treatment also reduced the glucose transporter 4 protein expression in both muscles. These results suggest that NOS plays a role in maintaining SIRT1 protein expression, muscle fiber composition and components of mitochondrial biogenesis in skeletal muscle. Key pointsNOS inhibition by L-NAME treatment decreased the SIRT1 protein expression in skeletal muscle.NOS inhibition induced the Type I to Type IIA fiber type transformation in soleus muscle.NOS inhibition reduced the components of mitochondrial biogenesis and glucose metabolism in skeletal muscle. | | 26336341
 |
Changes in mitochondrial function and mitochondria associated protein expression in response to 2-weeks of high intensity interval training.
Vincent, G; Lamon, S; Gant, N; Vincent, PJ; MacDonald, JR; Markworth, JF; Edge, JA; Hickey, AJ
Frontiers in physiology
6
51
2015
Show Abstract
High-intensity short-duration interval training (HIT) stimulates functional and metabolic adaptation in skeletal muscle, but the influence of HIT on mitochondrial function remains poorly studied in humans. Mitochondrial metabolism as well as mitochondrial-associated protein expression were tested in untrained participants performing HIT over a 2-week period.Eight males performed a single-leg cycling protocol (12 × 1 min intervals at 120% peak power output, 90 s recovery, 4 days/week). Muscle biopsies (vastus lateralis) were taken pre- and post-HIT. Mitochondrial respiration in permeabilized fibers, citrate synthase (CS) activity and protein expression of peroxisome proliferator-activated receptor gamma coactivator (PGC-1α) and respiratory complex components were measured.HIT training improved peak power and time to fatigue. Increases in absolute oxidative phosphorylation (OXPHOS) capacities and CS activity were observed, but not in the ratio of CCO to the electron transport system (CCO/ETS), the respiratory control ratios (RCR-1 and RCR-2) or mitochondrial-associated protein expression. Specific increases in OXPHOS flux were not apparent after normalization to CS, indicating that gross changes mainly resulted from increased mitochondrial mass.Over only 2 weeks HIT significantly increased mitochondrial function in skeletal muscle independently of detectable changes in mitochondrial-associated and mitogenic protein expression. | | 25759671
|
A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis.
Palamiuc, L; Schlagowski, A; Ngo, ST; Vernay, A; Dirrig-Grosch, S; Henriques, A; Boutillier, AL; Zoll, J; Echaniz-Laguna, A; Loeffler, JP; René, F
EMBO molecular medicine
7
526-46
2015
Show Abstract
Amyotrophic lateral sclerosis (ALS) is the most common fatal motor neuron disease in adults. Numerous studies indicate that ALS is a systemic disease that affects whole body physiology and metabolic homeostasis. Using a mouse model of the disease (SOD1(G86R)), we investigated muscle physiology and motor behavior with respect to muscle metabolic capacity. We found that at 65 days of age, an age described as asymptomatic, SOD1(G86R) mice presented with improved endurance capacity associated with an early inhibition in the capacity for glycolytic muscle to use glucose as a source of energy and a switch in fuel preference toward lipids. Indeed, in glycolytic muscles we showed progressive induction of pyruvate dehydrogenase kinase 4 expression. Phosphofructokinase 1 was inhibited, and the expression of lipid handling molecules was increased. This mechanism represents a chronic pathologic alteration in muscle metabolism that is exacerbated with disease progression. Further, inhibition of pyruvate dehydrogenase kinase 4 activity with dichloroacetate delayed symptom onset while improving mitochondrial dysfunction and ameliorating muscle denervation. In this study, we provide the first molecular basis for the particular sensitivity of glycolytic muscles to ALS pathology. | | 25820275
|
(-)-Epicatechin combined with 8 weeks of treadmill exercise is associated with increased angiogenic and mitochondrial signaling in mice.
Lee, I; Hüttemann, M; Kruger, A; Bollig-Fischer, A; Malek, MH
Frontiers in pharmacology
6
43
2015
Show Abstract
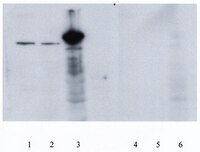
The purpose of this study was to conduct an 8 week endurance training program with and without (-)-epicatechin treatment and to determine whether there is a possible cumulative effect on protein markers of angiogenesis and mitochondrial biogenesis. Thirty-four 14-month old male mice (C57BL/6N) were randomized into four groups: control (C); (-)-epicatechin only ((-)-Epi); control with endurance training (CE); and (-)-epicatechin with endurance training ((-)-Epi-Ex). Mice in the training groups performed treadmill exercise for 8 weeks (5 × /week for 60 min/session), whereas mice in the (-)-epicatechin group received 1.0 mg/kg of body mass twice daily during the training period. At 8 weeks, distance ran on the treadmill increased by 46, 69, and 84% in the (-)-Epi, CE, and (-)-Epi-Ex groups, respectively compared to the control group (p less than 0.001 for all comparisons). Furthermore, the (-)-Epi-Ex group had significantly higher exercise capacity than the (-)-Epi and CE group. For angiogenic regulators, the (-)-Epi-Ex group had significantly higher VEGF-R2 protein expression with a concomitant reduction in TSP-1 protein expression than the exercise group. Interestingly, FoxO1 protein expression was significantly reduced for all three experimental groups compared to the control group. Protein markers such as PGC-1β and TFAM were significantly higher in the (-)-Epi-Ex group compared to the three other groups. These findings suggest that (-)-epicatechin treatment combined with 8 weeks of endurance training provide a cumulative effect on a number of angiogenic and mitochondrial signaling which functionally translates to enhanced exercise tolerance. | Western Blotting | 25821434
|
3,5-Diiodo-L-thyronine activates brown adipose tissue thermogenesis in hypothyroid rats.
Lombardi, A; Senese, R; De Matteis, R; Busiello, RA; Cioffi, F; Goglia, F; Lanni, A
PloS one
10
e0116498
2015
Show Abstract
3,5-Diiodo-l-thyronine (T2), a thyroid hormone derivative, is capable of increasing energy expenditure, as well as preventing high fat diet-induced overweight and related metabolic dysfunction. Most studies to date on T2 have been carried out on liver and skeletal muscle. Considering the role of brown adipose tissue (BAT) in energy and metabolic homeostasis, we explored whether T2 could activate BAT thermogenesis. Using euthyroid, hypothyroid, and T2-treated hypothyroid rats (all maintained at thermoneutrality) in morphological and functional studies, we found that hypothyroidism suppresses the maximal oxidative capacity of BAT and thermogenesis, as revealed by reduced mitochondrial content and respiration, enlarged cells and lipid droplets, and increased number of unilocular cells within the tissue. In vivo administration of T2 to hypothyroid rats activated BAT thermogenesis and increased the sympathetic innervation and vascularization of tissue. Likewise, T2 increased BAT oxidative capacity in vitro when added to BAT homogenates from hypothyroid rats. In vivo administration of T2 to hypothyroid rats enhanced mitochondrial respiration. Moreover, UCP1 seems to be a molecular determinant underlying the effect of T2 on mitochondrial thermogenesis. In fact, inhibition of mitochondrial respiration by GDP and its reactivation by fatty acids were greater in mitochondria from T2-treated hypothyroid rats than untreated hypothyroid rats. In vivo administration of T2 led to an increase in PGC-1α protein levels in nuclei (transient) and mitochondria (longer lasting), suggesting a coordinate effect of T2 in these organelles that ultimately promotes net activation of mitochondrial biogenesis and BAT thermogenesis. The effect of T2 on PGC-1α is similar to that elicited by triiodothyronine. As a whole, the data reported here indicate T2 is a thyroid hormone derivative able to activate BAT thermogenesis. | | 25658324
|
p53 is necessary for the adaptive changes in cellular milieu subsequent to an acute bout of endurance exercise.
Saleem, A; Carter, HN; Hood, DA
American journal of physiology. Cell physiology
306
C241-9
2014
Show Abstract
An acute bout of exercise activates downstream signaling cascades that ultimately result in mitochondrial biogenesis. In addition to inducing mitochondrial synthesis, exercise triggers the removal of damaged cellular material via autophagy and of dysfunctional mitochondria through mitophagy. Here, we investigated the necessity of p53 to the changes that transpire within the muscle upon an imposed metabolic and physiological challenge, such as a bout of endurance exercise. We randomly assigned wild-type (WT) and p53 knockout (KO) mice to control, acute exercise (AE; 90 min at 15 m/min), and AE + 3 h recovery (AER) groups and measured downstream alterations in markers of mitochondrial biogenesis, autophagy, and mitophagy. In the absence of p53, activation of p38 MAPK upon exercise was abolished, whereas CaMKII and AMP-activated protein kinase only displayed an attenuated enhancement in the AER group compared with WT mice. The translocation of peroxisome proliferator-activated receptor-γ coactivator-1 α to the nucleus was diminished and only observed in the AER group, and the subsequent increase in messenger RNA transcripts related to mitochondrial biogenesis with exercise and recovery was absent in the p53 KO animals. Whole-muscle autophagic and lysosomal markers did not respond to exercise, irrespective of the genotype of the exercised mice, with the exception of increased ubiquitination observed in KO mice with exercise. Markers of mitophagy were elevated in response to AE and AER conditions in both WT and p53 KO runners. The data suggest that p53 is important for the exercise-induced activation of mitochondrial synthesis and is integral in regulating autophagy during control conditions but not in response to exercise. | Western Blotting | 24284795
|
Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans.
Gouspillou, Gilles, et al.
FASEB J., 28: 1621-33 (2014)
2014
Show Abstract
Mitochondrial dysfunction is implicated in skeletal muscle atrophy and dysfunction with aging, with strong support for an increased mitochondrial-mediated apoptosis in sedentary rodent models. Whether this applies to aged human muscle is unknown, nor is it clear whether these changes are caused by sedentary behavior. Thus, we examined mitochondrial function [respiration, reactive oxygen species (ROS) emission, and calcium retention capacity (CRC)] in permeabilized myofibers obtained from vastus lateralis muscle biopsies of healthy physically active young (23.7±2.7 yr; mean±SD) and older (71.2±4.9 yr) men. Although mitochondrial ROS and maximal respiratory capacity were unaffected, the acceptor control ratio was reduced by 18% with aging, suggesting mild uncoupling of oxidative phosphorylation. CRC was reduced by 50% with aging, indicating sensitization of the mitochondrial permeability transition pore (mPTP) to apoptosis. Consistent with the mPTP sensitization, older muscles showed a 3-fold greater fraction of endonuclease G (a mitochondrial proapoptotic factor)-positive myonuclei. Aged muscles also had lower mitophagic potential, based on a 43% reduction in Parkin to the voltage-dependent anion channel (VDAC) protein ratio. Collectively, these results show that mitochondrial-mediated apoptotic signaling is increased in older human muscle and suggest that accumulation of dysfunctional mitochondria with exaggerated apoptotic sensitivity is due to impaired mitophagy. | | 24371120
|
PARK2/Parkin-mediated mitochondrial clearance contributes to proteasome activation during slow-twitch muscle atrophy via NFE2L1 nuclear translocation.
Furuya, N; Ikeda, S; Sato, S; Soma, S; Ezaki, J; Oliva Trejo, JA; Takeda-Ezaki, M; Fujimura, T; Arikawa-Hirasawa, E; Tada, N; Komatsu, M; Tanaka, K; Kominami, E; Hattori, N; Ueno, T
Autophagy
10
631-41
2014
Show Abstract
Skeletal muscle atrophy is thought to result from hyperactivation of intracellular protein degradation pathways, including autophagy and the ubiquitin-proteasome system. However, the precise contributions of these pathways to muscle atrophy are unclear. Here, we show that an autophagy deficiency in denervated slow-twitch soleus muscles delayed skeletal muscle atrophy, reduced mitochondrial activity, and induced oxidative stress and accumulation of PARK2/Parkin, which participates in mitochondrial quality control (PARK2-mediated mitophagy), in mitochondria. Soleus muscles from denervated Park2 knockout mice also showed resistance to denervation, reduced mitochondrial activities, and increased oxidative stress. In both autophagy-deficient and Park2-deficient soleus muscles, denervation caused the accumulation of polyubiquitinated proteins. Denervation induced proteasomal activation via NFE2L1 nuclear translocation in control mice, whereas it had little effect in autophagy-deficient and Park2-deficient mice. These results suggest that PARK2-mediated mitophagy plays an essential role in the activation of proteasomes during denervation atrophy in slow-twitch muscles. | | 24451648
|
The relationship between muscle fiber type-specific PGC-1α content and mitochondrial content varies between rodent models and humans.
Gouspillou, G; Sgarioto, N; Norris, B; Barbat-Artigas, S; Aubertin-Leheudre, M; Morais, JA; Burelle, Y; Taivassalo, T; Hepple, RT
PloS one
9
e103044
2014
Show Abstract
PGC-1α regulates critical processes in muscle physiology, including mitochondrial biogenesis, lipid metabolism and angiogenesis. Furthermore, PGC-1α was suggested as an important regulator of fiber type determination. However, whether a muscle fiber type-specific PGC-1α content exists, whether PGC-1α content relates to basal levels of mitochondrial content, and whether such relationships are preserved between humans and classically used rodent models are all questions that have been either poorly addressed or never investigated. To address these issues, we investigated the fiber type-specific content of PGC-1α and its relationship to basal mitochondrial content in mouse, rat and human muscles using in situ immunolabeling and histochemical methods on muscle serial cross-sections. Whereas type IIa fibers exhibited the highest PGC-1α in all three species, other fiber types displayed a hierarchy of type IIxgreater than Igreater than IIb in mouse, type I = IIxgreater than IIb in rat, and type IIxgreater than I in human. In terms of mitochondrial content, we observed a hierarchy of IIagreater than IIxgreater than Igreater than IIb in mouse, IIa greater than Igreater than IIxgreater than IIb in rat, and Igreater than IIagreater than IIx in human skeletal muscle. We also found in rat skeletal muscle that type I fibers displayed the highest capillarization followed by type IIa greater than IIxgreater than IIb. Finally, we found in human skeletal muscle that type I fibers display the highest lipid content, followed by type IIagreater than IIx. Altogether, our results reveal that (i) the fiber type-specific PGC-1α and mitochondrial contents were only matched in mouse, (ii) the patterns of PGC-1α and mitochondrial contents observed in mice and rats do not correspond to that seen in humans in several respects, and (iii) the classical phenotypes thought to be regulated by PGC-1α do not vary exclusively as a function of PGC-1α content in rat and human muscles. | | 25121500
|
Dynamic partnership between TFIIH, PGC-1α and SIRT1 is impaired in trichothiodystrophy.
Traboulsi, H; Davoli, S; Catez, P; Egly, JM; Compe, E
PLoS genetics
10
e1004732
2014
Show Abstract
The expression of protein-coding genes requires the selective role of many transcription factors, whose coordinated actions remain poorly understood. To further grasp the molecular mechanisms that govern transcription, we focused our attention on the general transcription factor TFIIH, which gives rise, once mutated, to Trichothiodystrophy (TTD), a rare autosomal premature-ageing disease causing inter alia, metabolic dysfunctions. Since this syndrome could be connected to transcriptional defects, we investigated the ability of a TTD mouse model to cope with food deprivation, knowing that energy homeostasis during fasting involves an accurate regulation of the gluconeogenic genes in the liver. Abnormal amounts of gluconeogenic enzymes were thus observed in TTD hepatic parenchyma, which was related to the dysregulation of the corresponding genes. Strikingly, such gene expression defects resulted from the inability of PGC1-α to fulfill its role of coactivator. Indeed, extensive molecular analyses unveiled that wild-type TFIIH cooperated in an ATP-dependent manner with PGC1-α as well as with the deacetylase SIRT1, thereby contributing to the PGC1-α deacetylation by SIRT1. Such dynamic partnership was, however, impaired when TFIIH was mutated, having as a consequence the disruption of PGC1-α recruitment to the promoter of target genes. Therefore, besides a better understanding of the etiology of TFIIH-related disease, our results shed light on the synergistic relationship that exist between different types of transcription factors, which is necessary to properly regulate the expression of protein coding genes. | | 25340339
|