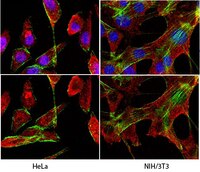
B-cell precursor acute lymphoblastic leukemia and stromal cells communicate through Galectin-3.
Fei, F; Joo, EJ; Tarighat, SS; Schiffer, I; Paz, H; Fabbri, M; Abdel-Azim, H; Groffen, J; Heisterkamp, N
Oncotarget
6
11378-94
2015
Mostrar resumen
The molecular interactions between B-cell precursor acute lymphoblastic leukemia (pre-B ALL) cells and stromal cells in the bone marrow that provide microenvironmentally-mediated protection against therapeutic drugs are not well-defined. Galectin-3 (Lgals3) is a multifunctional galactose-binding lectin with reported location in the nucleus, cytoplasm and extracellular space in different cell types. We previously reported that ALL cells co-cultured with stroma contain high levels of Galectin-3. We here establish that, in contrast to more mature B-lineage cancers, Galectin-3 detected in and on the ALL cells originates from stromal cells, which express it on their surface, secrete it as soluble protein and also in exosomes. Soluble and stromal-bound Galectin-3 is internalized by ALL cells, transported to the nucleus and stimulates transcription of endogenous LGALS3 mRNA. When human and mouse ALL cells develop tolerance to different drugs while in contact with protective stromal cells, Galectin-3 protein levels are consistently increased. This correlates with induction of Galectin-3 transcription in the ALL cells. Thus Galectin-3 sourced from stroma becomes supplemented by endogenous Galectin-3 production in the pre-B ALL cells that are under continuous stress from drug treatment. Our data suggest that stromal Galectin-3 may protect ALL cells through auto-induction of Galectin-3 mRNA and tonic NFκB pathway activation. Since endogenously synthesized Galectin-3 protects pre-B ALL cells against drug treatment, we identify Galectin-3 as one possible target to counteract the protective effects of stroma. | | | 25869099
 |
The mTORC1/4E-BP pathway coordinates hemoglobin production with L-leucine availability.
Chung, J; Bauer, DE; Ghamari, A; Nizzi, CP; Deck, KM; Kingsley, PD; Yien, YY; Huston, NC; Chen, C; Schultz, IJ; Dalton, AJ; Wittig, JG; Palis, J; Orkin, SH; Lodish, HF; Eisenstein, RS; Cantor, AB; Paw, BH
Science signaling
8
ra34
2015
Mostrar resumen
In multicellular organisms, the mechanisms by which diverse cell types acquire distinct amino acids and how cellular function adapts to their availability are fundamental questions in biology. We found that increased neutral essential amino acid (NEAA) uptake was a critical component of erythropoiesis. As red blood cells matured, expression of the amino acid transporter gene Lat3 increased, which increased NEAA import. Inadequate NEAA uptake by pharmacologic inhibition or RNAi-mediated knockdown of LAT3 triggered a specific reduction in hemoglobin production in zebrafish embryos and murine erythroid cells through the mTORC1 (mammalian target of rapamycin complex 1)/4E-BP (eukaryotic translation initiation factor 4E-binding protein) pathway. CRISPR-mediated deletion of members of the 4E-BP family in murine erythroid cells rendered them resistant to mTORC1 and LAT3 inhibition and restored hemoglobin production. These results identify a developmental role for LAT3 in red blood cells and demonstrate that mTORC1 serves as a homeostatic sensor that couples hemoglobin production at the translational level to sufficient uptake of NEAAs, particularly L-leucine. | Western Blotting | | 25872869
|
Loss of F-box only protein 2 (Fbxo2) disrupts levels and localization of select NMDA receptor subunits, and promotes aberrant synaptic connectivity.
Atkin, G; Moore, S; Lu, Y; Nelson, RF; Tipper, N; Rajpal, G; Hunt, J; Tennant, W; Hell, JW; Murphy, GG; Paulson, H
The Journal of neuroscience : the official journal of the Society for Neuroscience
35
6165-78
2015
Mostrar resumen
NMDA receptors (NMDARs) play an essential role in some forms of synaptic plasticity, learning, and memory. Therefore, these receptors are highly regulated with respect to their localization, activation, and abundance both within and on the surface of mammalian neurons. Fundamental questions remain, however, regarding how this complex regulation is achieved. Using cell-based models and F-box Only Protein 2 (Fbxo2) knock-out mice, we found that the ubiquitin ligase substrate adaptor protein Fbxo2, previously reported to facilitate the degradation of the NMDAR subunit GluN1 in vitro, also functions to regulate GluN1 and GluN2A subunit levels in the adult mouse brain. In contrast, GluN2B subunit levels are not affected by the loss of Fbxo2. The loss of Fbxo2 results in greater surface localization of GluN1 and GluN2A, together with increases in the synaptic markers PSD-95 and Vglut1. These synaptic changes do not manifest as neurophysiological differences or alterations in dendritic spine density in Fbxo2 knock-out mice, but result instead in increased axo-dendritic shaft synapses. Together, these findings suggest that Fbxo2 controls the abundance and localization of specific NMDAR subunits in the brain and may influence synapse formation and maintenance. | | | 25878288
|
Comparison of the metabolic activation of environmental carcinogens in mouse embryonic stem cells and mouse embryonic fibroblasts.
Krais, AM; Mühlbauer, KR; Kucab, JE; Chinbuah, H; Cornelius, MG; Wei, QX; Hollstein, M; Phillips, DH; Arlt, VM; Schmeiser, HH
Toxicology in vitro : an international journal published in association with BIBRA
29
34-43
2015
Mostrar resumen
We compared mouse embryonic stem (ES) cells and fibroblasts (MEFs) for their ability to metabolically activate the environmental carcinogens benzo[a]pyrene (BaP), 3-nitrobenzanthrone (3-NBA) and aristolochic acid I (AAI), measuring DNA adduct formation by (32)P-postlabelling and expression of xenobiotic-metabolism genes by quantitative real-time PCR. At 2 μM, BaP induced Cyp1a1 expression in MEFs to a much greater extent than in ES cells and formed 45 times more adducts. Nqo1 mRNA expression was increased by 3-NBA in both cell types but induction was higher in MEFs, as was adduct formation. For AAI, DNA binding was over 450 times higher in MEFs than in ES cells, although Nqo1 and Cyp1a1 transcriptional levels did not explain this difference. We found higher global methylation of DNA in ES cells than in MEFs, which suggests higher chromatin density and lower accessibility of the DNA to DNA damaging agents in ES cells. However, AAI treatment did not alter DNA methylation. Thus mouse ES cells and MEFs have the metabolic competence to activate a number of environmental carcinogens, but MEFs have lower global DNA methylation and higher metabolic capacity than mouse ES cells. | | | 25230394
|
Inhibitory effect of statins on inflammation-related pathways in human abdominal aortic aneurysm tissue.
Yoshimura, K; Nagasawa, A; Kudo, J; Onoda, M; Morikage, N; Furutani, A; Aoki, H; Hamano, K
International journal of molecular sciences
16
11213-28
2015
Mostrar resumen
HMG-CoA (3-hydroxy-3-methylglutaryl-coenzyme A) reductase inhibitors (statins) have been suggested to attenuate abdominal aortic aneurysm (AAA) growth. However, the effects of statins in human AAA tissues are not fully elucidated. The aim of this study was to investigate the direct effects of statins on proinflammatory molecules in human AAA walls in ex vivo culture. Simvastatin strongly inhibited the activation of nuclear factor (NF)-κB induced by tumor necrosis factor (TNF)-α in human AAA walls, but showed little effect on c-jun N-terminal kinase (JNK) activation. Simvastatin, as well as pitavastatin significantly reduced the secretion of matrix metalloproteinase (MMP)-9, monocyte chemoattractant protein (MCP)-2 and epithelial neutrophil-activating peptide (CXCL5) under both basal and TNF-α-stimulated conditions. Similar to statins, the Rac1 inhibitor NSC23766 significantly inhibited the activation of NF-κB, accompanied by a decreased secretion of MMP-9, MCP-2 and CXCL5. Moreover, the effect of simvastatin and the JNK inhibitor SP600125 was additive in inhibiting the secretion of MMP-9, MCP-2 and CXCL5. These findings indicate that statins preferentially inhibit the Rac1/NF-κB pathway to suppress MMP-9 and chemokine secretion in human AAA, suggesting a mechanism for the potential effect of statins in attenuating AAA progression. | | | 25993292
|
Dietary Vitamin D3 Restriction Exacerbates Disease Pathophysiology in the Spinal Cord of the G93A Mouse Model of Amyotrophic Lateral Sclerosis.
Moghimi, E; Solomon, JA; Gianforcaro, A; Hamadeh, MJ
PloS one
10
e0126355
2015
Mostrar resumen
Dietary vitamin D3 (D3) restriction reduces paw grip endurance and motor performance in G93A mice, and increases inflammation and apoptosis in the quadríceps of females. ALS, a neuromuscular disease, causes progressive degeneration of motor neurons in the brain and spinal cord.We analyzed the spinal cords of G93A mice following dietary D3 restriction at 2.5% the adequate intake (AI) for oxidative damage (4-HNE, 3-NY), antioxidant enzymes (SOD2, catalase, GPx1), inflammation (TNF-α, IL-6, IL-10), apoptosis (bax/bcl-2 ratio, cleaved/pro-caspase 3 ratio), neurotrophic factor (GDNF) and neuron count (ChAT, SMI-36/SMI-32 ratio).Beginning at age 25 d, 42 G93A mice were provided food ad libitum with either adequate (AI;1 IU D3/g feed; 12 M, 11 F) or deficient (DEF; 0.025 IU D3/g feed; 10 M, 9 F) D3. At age 113 d, the spinal cords were analyzed for protein content. Differences were considered significant at P ≤ 0.10, since this was a pilot study.DEF mice had 16% higher 4-HNE (P = 0.056), 12% higher GPx1 (P = 0.057) and 23% higher Bax/Bcl2 ratio (P = 0.076) vs. AI. DEF females had 29% higher GPx1 (P = 0.001) and 22% higher IL-6 (P = 0.077) vs. AI females. DEF males had 23% higher 4-HNE (P = 0.066) and 18% lower SOD2 (P = 0.034) vs. AI males. DEF males had 27% lower SOD2 (P = 0.004), 17% lower GPx1 (P = 0.070), 29% lower IL-6 (P = 0.023) and 22% lower ChAT (P = 0.082) vs. DEF females.D3 deficiency exacerbates disease pathophysiology in the spinal cord of G93A mice, the exact mechanisms are sex-specific. This is in accord with our previous results in the quadriceps, as well as functional and disease outcomes. | | | 26020962
|
CSB interacts with SNM1A and promotes DNA interstrand crosslink processing.
Iyama, T; Lee, SY; Berquist, BR; Gileadi, O; Bohr, VA; Seidman, MM; McHugh, PJ; Wilson, DM
Nucleic acids research
43
247-58
2015
Mostrar resumen
Cockayne syndrome (CS) is a premature aging disorder characterized by photosensitivity, impaired development and multisystem progressive degeneration, and consists of two strict complementation groups, A and B. Using a yeast two-hybrid approach, we identified the 5'-3' exonuclease SNM1A as one of four strong interacting partners of CSB. This direct interaction was confirmed using purified recombinant proteins-with CSB able to modulate the exonuclease activity of SNM1A on oligonucleotide substrates in vitro-and the two proteins were shown to exist in a common complex in human cell extracts. CSB and SNM1A were also found, using fluorescently tagged proteins in combination with confocal microscopy and laser microirradiation, to be recruited to localized trioxsalen-induced ICL damage in human cells, with accumulation being suppressed by transcription inhibition. Moreover, SNM1A recruitment was significantly reduced in CSB-deficient cells, suggesting coordination between the two proteins in vivo. CSB-deficient neural cells exhibited increased sensitivity to DNA crosslinking agents, particularly, in a non-cycling, differentiated state, as well as delayed ICL processing as revealed by a modified Comet assay and γ-H2AX foci persistence. The results indicate that CSB coordinates the resolution of ICLs, possibly in a transcription-associated repair mechanism involving SNM1A, and that defects in the process could contribute to the post-mitotic degenerative pathologies associated with CS. | | | 25505141
|
HMGB1 facilitates repair of mitochondrial DNA damage and extends the lifespan of mutant ataxin-1 knock-in mice.
Ito, H; Fujita, K; Tagawa, K; Chen, X; Homma, H; Sasabe, T; Shimizu, J; Shimizu, S; Tamura, T; Muramatsu, S; Okazawa, H
EMBO molecular medicine
7
78-101
2015
Mostrar resumen
Mutant ataxin-1 (Atxn1), which causes spinocerebellar ataxia type 1 (SCA1), binds to and impairs the function of high-mobility group box 1 (HMGB1), a crucial nuclear protein that regulates DNA architectural changes essential for DNA damage repair and transcription. In this study, we established that transgenic or virus vector-mediated complementation with HMGB1 ameliorates motor dysfunction and prolongs lifespan in mutant Atxn1 knock-in (Atxn1-KI) mice. We identified mitochondrial DNA damage repair by HMGB1 as a novel molecular basis for this effect, in addition to the mechanisms already associated with HMGB1 function, such as nuclear DNA damage repair and nuclear transcription. The dysfunction and the improvement of mitochondrial DNA damage repair functions are tightly associated with the exacerbation and rescue, respectively, of symptoms, supporting the involvement of mitochondrial DNA quality control by HMGB1 in SCA1 pathology. Moreover, we show that the rescue of Purkinje cell dendrites and dendritic spines by HMGB1 could be downstream effects. Although extracellular HMGB1 triggers inflammation mediated by Toll-like receptor and receptor for advanced glycation end products, upregulation of intracellular HMGB1 does not induce such side effects. Thus, viral delivery of HMGB1 is a candidate approach by which to modify the disease progression of SCA1 even after the onset. | | | 25510912
|
Krüppel-like factor 6 regulates mitochondrial function in the kidney.
Mallipattu, SK; Horne, SJ; D'Agati, V; Narla, G; Liu, R; Frohman, MA; Dickman, K; Chen, EY; Ma'ayan, A; Bialkowska, AB; Ghaleb, AM; Nandan, MO; Jain, MK; Daehn, I; Chuang, PY; Yang, VW; He, JC
The Journal of clinical investigation
125
1347-61
2015
Mostrar resumen
Maintenance of mitochondrial structure and function is critical for preventing podocyte apoptosis and eventual glomerulosclerosis in the kidney; however, the transcription factors that regulate mitochondrial function in podocyte injury remain to be identified. Here, we identified Krüppel-like factor 6 (KLF6), a zinc finger domain transcription factor, as an essential regulator of mitochondrial function in podocyte apoptosis. We observed that podocyte-specific deletion of Klf6 increased the susceptibility of a resistant mouse strain to adriamycin-induced (ADR-induced) focal segmental glomerulosclerosis (FSGS). KLF6 expression was induced early in response to ADR in mice and cultured human podocytes, and prevented mitochondrial dysfunction and activation of intrinsic apoptotic pathways in these podocytes. Promoter analysis and chromatin immunoprecipitation studies revealed that putative KLF6 transcriptional binding sites are present in the promoter of the mitochondrial cytochrome c oxidase assembly gene (SCO2), which is critical for preventing cytochrome c release and activation of the intrinsic apoptotic pathway. Additionally, KLF6 expression was reduced in podocytes from HIV-1 transgenic mice as well as in renal biopsies from patients with HIV-associated nephropathy (HIVAN) and FSGS. Together, these findings indicate that KLF6-dependent regulation of the cytochrome c oxidase assembly gene is critical for maintaining mitochondrial function and preventing podocyte apoptosis. | | | 25689250
|
Behavior-linked FoxP2 regulation enables zebra finch vocal learning.
Heston, JB; White, SA
The Journal of neuroscience : the official journal of the Society for Neuroscience
35
2885-94
2015
Mostrar resumen
Mutations in the FOXP2 transcription factor cause an inherited speech and language disorder, but how FoxP2 contributes to learning of these vocal communication signals remains unclear. FoxP2 is enriched in corticostriatal circuits of both human and songbird brains. Experimental knockdown of this enrichment in song control neurons of the zebra finch basal ganglia impairs tutor song imitation, indicating that adequate FoxP2 levels are necessary for normal vocal learning. In unmanipulated birds, vocal practice acutely downregulates FoxP2, leading to increased vocal variability and dynamic regulation of FoxP2 target genes. To determine whether this behavioral regulation is important for song learning, here, we used viral-driven overexpression of FoxP2 to counteract its downregulation. This manipulation disrupted the acute effects of song practice on vocal variability and caused inaccurate song imitation. Together, these findings indicate that dynamic behavior-linked regulation of FoxP2, rather than absolute levels, is critical for vocal learning. | | | 25698728
|